























-
-

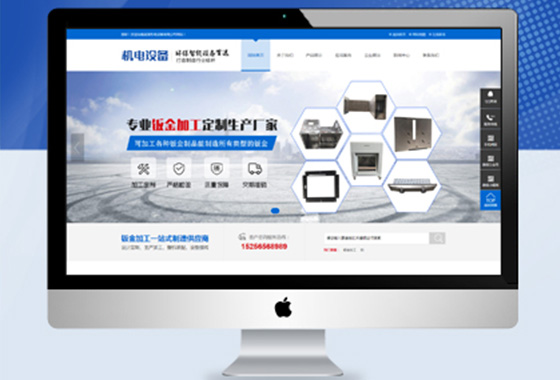
随县福润养殖项目网上二次公示美女操鸡巴粉嫩嫩av
넶66 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
-
-
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
-
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
- 2024-04-19
-
-
2024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-192024-04-19ꂀ 虚拟主机知识大全2024-04-192024-04-192024-04-19



























ꁲ









